
1. Introduction
1.1 Definition
The Pharmaceutical Quality by Design (QbD) is a systematic approach to development that
begins with predefined objectives and emphasizes product and process understanding and
process control, based on sound science and quality risk management. Quality by Design
(QbD) is emerging to enhance the assurance of safe, effective drug supply to the consumer
and also offers promise to significantly improve manufacturing quality performance.

1.2 Regulatory Requirements:
The concept of “Quality by Design” (QbD) was defined as an approach which covers a better
scientific understanding of critical process and product qualities, designing controls and tests
based on the scientific limits of understanding during the development phase and using the
knowledge obtained during the life-cycle of the product to work on a constant improvement
environment. QbD describes a pharmaceutical development approach referring to
formulation design and development and manufacturing processes to maintain the prescribed
product quality. Guidelines and mathematical models are used to ensure the establishment
and use of the knowledge on the subject in an independent and integrated way.
Within this vision, the key framework guidance documents ICH Q8 Pharmaceutical
Development and ICH Q9 Quality Risk Management were published in 2005 and ICH Q10
Pharmaceutical Quality System followed these documents in 2008. Q8 Pharmaceutical
Development focuses on the content of the Module 3.2.P.2 of the Common Technical
Document (CTD) and promotes the concept of QbD. Final guideline Q8(R)2 was published
in 2008. It supports knowledge gained through the lifecycle of a product and using scientific
approaches and quality risk management principles. Within the scope of Q8 Pharmaceutical
Development, an important step in the QbD approach to the development of drug products
requires a distinction between critical and non-critical product attributes and process
parameters.
Additionally, the quality target product profile (QTTP) is defined as a summary of the quality
characteristics or attributes of a product that ideally will be achieved and is related to quality,
safety and efficacy, considering the route of administration, dosage form, bioavailability,
strength, and stability.
The ICH guideline Q9 Quality Risk Management defines risk and offers a systematic
approach to quality risk management via describing how to conduct risk assessments and to
manage the risks defined providing guidance on the principles and some of the tools of
quality risk management. It can be a guide as a resource document that is independent of, yet
supporting, other ICH Quality documents and complementing existing quality practices,
requirements, standards, and guidelines within the pharmaceutical industry and regulatory
framework.
The ICH guideline Q10 describes a model for an effective pharmaceutical quality system
that is based on International Standards Organization (ISO) quality concepts, includes
applicable GMP regulations and complements ICH Q8 and ICH Q9, and is applicable for a
lifecycle of a product.
This guideline focuses on regulating the quality management systems of drug manufacturers;
where by any changes to manufacturing processes would be managed by appropriate change
control procedures have been developed.
Although the initial focus of the science and risk based agenda was linked primarily to drug
product, greater emphasis is now being placed on drug substance with the evolution of ICH
Q11 dedicated to the manufacture of drug raw materials. Recent regulatory initiatives such as
Quality-by-Design provided unprecedented opportunities to go beyond what was done in the
past. The guideline focuses on development and manufacturing process of both chemical and
biotechnological/biological drug substances and is intended to provide guidance in the scope
of ICH Guideline Q6A and Q6B. If the principles described in the ICH Q8, Q9 and Q10
guidance documents are implemented together in a holistic manner, then an effective system
that emphasizes a harmonized science and risk-based approach to product development and
maintenance is in place. This provides an even greater (quality) assurance that the patient
will receive product that meets the critical quality attributes (CQA).
1.3 Key Characteristics of QbD
A tool for focused & efficient drug development.
Dynamic and systematic process.
Relies on the concept that Quality can be built in as a continuum.
It is applicable to Drug Product and Drug Substance development (chemicals / biologics).
It is applicable to analytical methods.
Can implement partially or totally.
Can be used at any time in the life cycle of the Drug.
Always encouraged by Regulators.
1.4 Advantages of QbD
Benefits for Industry:
Better understanding of the process.
Less batch failure.
More efficient and effective control of change.
Return on investment / cost savings.
1.5 Factors impeding implementation on A QbD
Complete understanding of A QbD is still lacking in the pharmaceutical industry.
Proper definitions of the MODR, ATP, analytical method control strategy, and method
performance criteria and other elements of AQbD must be given. Global harmonization of
these terminologies is a must.
Revision of ICH guideline Q2 (R1) requires revision to include AQbD elements.
Additional guidelines need to be developed for implementation of AQbD.
Analysts must learn new tools and skills.
A consistent worldwide approach is required for this initiative to be effective.
1.6 Additional opportunities:
An enhance QbD approach to pharmaceutical development provides opportunities for
more flexible regulatory approaches. Ex: Manufacturing changes within the approved
design space without further regulatory review.
Reduction of post-approval submissions.
Better innovation due to the ability to improve processes without resubmission to the
FDA when remaining in the Design Space.
More efficient technology transfer to manufacturing.
Greater regulator confidence of robust products.
Risk-based approach and identification.
Innovative process validation approaches.
Less intense regulatory oversight and less post-approval submissions.
For the consumer, greater drug consistency.
More drug availability and less recall.
Improved yields, lower cost, less investigations, reduced testing, etc.
Time to market reductions: from 12 to 6 years realized by amongst others.
First time right: lean assets management.
Continuous improvement over the total product life cycle (i.e. controlled, patient guided
variability).
Absence of design freeze (no variation issues).
Less validation burden.
Real time controls (less batch controls).
Realistic risk perceptions.
Contributes substantially to realize the better, cheaper and safer mandate.
1.7 Implementation of QbD
A framework for applying QbD to analytical methods: QbD can be applied equally to
processes and analytical. It displays how QbD for methods, analytical methods are driven by
the overall process.
Method performance requirements (Design intent): Fundamental to any method
development is being clear about the design intent of the method (i.e., the criteria that must
be met). Method-performance criteria and method-operational intent are two important
aspects of this design intent.
Method performance criteria: These criteria are driven by an understanding of the
process monitoring and control requirements; that is, the process critical quality attributes
(CQAs) and specification limits. CQAs are identified, through a thorough understanding of
those characteristics of a drug substance or a drug product that may need to be controlled to
ensure the safety or efficacy of a product. For methods measuring these CQAs, criteria, such
as the following would need to be met. Precision—the need for method variability to be a
small proportion of the specification. Selectivity—being clear on which impurities actually
need to be monitored at each step in a process and ensuring adequate discrimination between
them. Sensitivity-ensuring the method is sufficiently sensitive relative to the specification
limit. PAT methods often meet the criteria above in a different way from traditional endpoint
testing methods such as high-performance liquid chromatography (HPLC). Selectivity,
for example, may be achieved through a multivariate model as opposed to the resolution
between adjacent peaks in an HPLC method. For these methods, precision could be
demonstrated by checking the predictive validity of the model.
Method operational intent: These criteria address the aspects of the method that are
required to facilitate ease of use in routine operation (e.g., analysis time, acceptable solvents,
and available equipment). Opportunities for the implementation of improved or new
technology also may be identified. These criteria can be generated by performing an analysis
of the voice of the customer (VoC) (i.e., the aspects of a method that are considered important for the quality control laboratories within manufacturing where the commercial
methods will be operated)
Method development (design selection): Fundamental to design selection is the methoddevelopment
phase. To develop a QbD method, the method performance criteria must be
understood as well as the desired operational intent that the eventual end user would wish to
see in the method.
1.8 QbD development process includes:

2. Elements of QbD
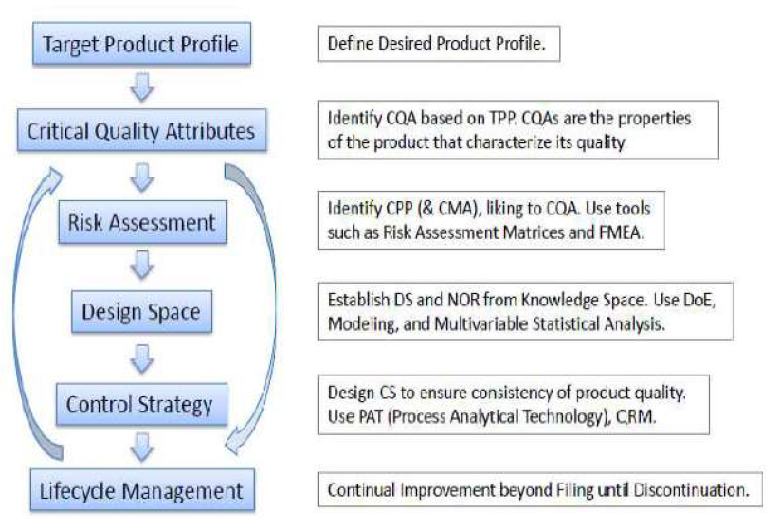
2.1 Quality target product profile (QTPP)
It is defined as a “prospective and dynamic summary of the quality characteristics of a
drug product that ideally will be achieved to ensure that the desired quality, andthus the
safety and efficacy, of a drug product is realized”. Basically it is a tool for setting the strategy
for drug development.
Recently QTTP is widely used in development planning, clinical and commercial
decision making, regulatory agency interactions, and risk management.
It is the quality characteristics that the drug product should possess in order to
reproducibly deliver the therapeutic benefit promised in the label.
The QTTP guides formulation scientists to establish formulation strategies and keep the
formulation effort focused and efficient.
QTPP is related to identity, assay, dosage form, purity, stability in the label.
The quality target product profile forms the basis of design for the development of the
product. Considerations for the quality target product profile could include:
Intended use in clinical setting, route of administration, dosage form, delivery systems;
Dosage strength(s);
Container closure system;
Therapeutic moiety release or delivery and attributes affecting pharmacokinetic
characteristics (e.g., dissolution, aerodynamic performance) appropriate to the drug product
dosage form being developed;
Drug product quality criteria (e.g., sterility, purity, stability and drug release) appropriate
for the intended marketed product.
2.2 Critical Quality Attributes (CQA)
Once TPQP has been identified, the next step is to identify the relevant CQAs.
A CQA is a physical, chemical, biological,or microbiological property or characteristic
that should be within an appropriate limit, range, or distribution to ensure the desired product
quality. CQAs are generally associated with the drug substance, excipients, intermediates (inprocess
materials) and drug product.
CQAs of solid oral dosage forms are typically those aspects affecting product purity,
strength, drug release and stability. CQAs for other delivery systems can additionally include
more product specific aspects, such as aerodynamic properties for inhaled products, sterility
for parenterals, and adhesion properties for transdermal patches. For drug substances, raw
materials and intermediates, the CQAs can additionally include those properties (e.g., particle
size distribution, bulk density) that affect drug product CQAs.
Potential drug product CQAs derived from the quality target product profile and/or prior
knowledge is used to guide the product and process development. The list of potential CQAs
can be modified when the formulation and manufacturing process are selected and as product
knowledge and process understanding increase.
Quality risk management can be used to prioritize the list of potential CQAs for
subsequent evaluation. Relevant CQAs can be identified by an iterative process of quality
risk management and experimentation that assesses the extent to which their variation can
have an impact on the quality of the drug product.
The use of robust risk assessment methods for identification of CQAs is novel to the QbD
paradigm CQAs of solid oral dosage forms are typically those aspects affecting product
purity, strength, drug release and stability.
CQAs for other delivery systems can additionally include more product specific aspects,
such as aerodynamic properties for inhaled products, sterility for parenteral, and adhesion
properties for transdermal patches.
2.3 Critical Process Parameter
Critical process parameters (CPPs) are defined as “parameters whose variability have an
impact on a CQA and therefore should be monitored or controlled to ensure the process
produces the desired quality.
A parameter is critical when a realistic change in that parameter can cause the product to
fail to meet the QTPP. Thus, whether a parameter is critical or not depends on how large of a
change one is willing to consider.
Process robustness is defined as the ability of a process to demonstrate acceptable quality
and performance and tolerate variability in inputs at the same time. To demonstrate the
reproducibility and consistency of a process, process capability should be studied. Process
capability is a statistical measure of the inherent process variability for a given
characteristics.
The most widely accepted formula for process capability is six sigma.
Process capability index is the value of the tolerance specified for a particular
characteristic divided by the process capability, which is defined as follows:
Process capability index (CpK) = Upper limit of specification - Lower limit of specification
6 standard deviation
If the CpK is significantly greater than one, the process is defined capable. If the process
capability is low, there are five step procedures to progressively reduce the variability of the
process.
These five steps are:
I. Define: The intended improvement should be clearly stated.
II. Measure: The critical product performance attributes should be measured to see if they are
out of specification and used to the sigma level of the process.
III. Analyze: When the sigma level is below the target, steps should be taken to increase it,
starting by identifying the most significant causes of the excessive variability.
IV. Improve: The process should be redesigned and/ or process controls should be
incorporated to eliminate or attenuate the significant toot causes of variance.
V. Control: The improved manufacturing process should be evaluated and maintained.
2.4 Risk assessment
Quality risk management is a systematic process for the assessment, control,
communication and review of risks to the quality of the drug (medicinal) product across the
product lifecycle.
The initial list of potential parameters which can affect CQAs can be quite extensive but
can be reduced and prioritized by quality risk assessment (QRA).
QRA is a science based process that can aid identification of CPPs and thus eliminating
risk, resulting in high confidence that the analytical method will meet the QTTP under all
conditions of use.
Thus, a large number of parameters can actually be safely eliminated by use of QRA
tools, for example failure mode effects analysis (FMEA) and Ishikawa diagrams on the basis
of prior knowledge and initial experimentation.
In FMEA the variables are ranked on the basis of the likelihood failure will occur
(probability), affect on the pharmaceutical results (severity), and difficulty of detection
(detectability), resulting in a risk priority number (RPN).
Factors with an RPN above a cut-off level can then be evaluated by subsequent studies
whereas factors with a lower RPN can be eliminated from further study.
Ishikawa diagrams segregate risks into different categories, for example those associated
with instrumentation, materials, methods, measurements, laboratory climate, and human
factors.
2.5 Design Space
The ICH Q8 (R2) states that the design space is multidimensional combination and
interaction of input variables (e.g., material attributes) and process parameters that have been
demonstrated to provide assurance of quality.
Working within the design space is not considered as a change. Movement out of the
design space is considered to be a change and would normally initiate a regulatory post
approval change process.
Design space may be constructed for a single unit operation, multiple unit operations, or
for the entire process.
Though according to FDA guideline, defining design space is optional since the product
and process understanding can be established without a formal design space, nevertheless,
such approach can assist to better understanding and attain overall control of a system.
Design space is proposed by the applicant and is subject to regulatory assessment and
approval.
It describes the multivariate functional relationships between CQAs and the CPPs that
impact them, and should include their linkage to or across unit operations.
Such relationships are arrived at by iterative application of risk assessment and
experimental design, modeling, as well as the use of literature and prior experience.
Methods for determining design space included: one-variable-at-a-time experiments,
statistically designed experiments, and modeling approaches.
Methods for presenting design space included graphs (surface-response curves and
contour plots), linear combination of parameter ranges, equations, and models.
Design space is potentially scale and equipment dependent, the design space determined
on the laboratory scale may not be relevant to the process at the commercial scale. Therefore,
design-space verification at the commercial scale becomes essential unless it is confirmed
that the design space is scale-independent.
2.6 Control Strategy
The ability to evaluate and ensure the quality of in-process and/or final product based on
process data which typically include a valid combination of measured material attributes
and process controls. ICH Q8 (R2).
Control strategy is defined as “a planned set of controls, derived from current product and
process understanding that assures process performance and product quality”.
The control strategy in the QbD paradigm is established via risk assessment that takes
into account the criticality of the CQA and process capability.
DOE results can help identify optimal conditions, the critical factors that most influence
CQAs and those who do not, as well as details such as the existence of interactions and
synergies between factors. The control strategy can include the following elements:
procedural controls, in-process controls, lot release testing, process monitoring,
characterization testing, comparability testing and stability testing.
Particularly, the control strategy may include:
Control of raw material attributes (e.g., drug substance, excipients and primary
packaging materials) based on an understanding of their impact on process-ability
or product quality.
Product specifications
Procedural controls
Facility controls such as utilities, environmental systems and operating conditions
Controls for unit operations that have an impact on downstream processing or
end-product quality (e.g. the impact of drying on degradation, particle size
distribution of the granulate on dissolution).
2.7 Life Cycle Management
In the QbD paradigm, process changes within the design space will not require review or
approval.
Therefore, process improvements during the product life cycle with regard to process
consistency and throughput could take place with fewer post approval submissions.
In addition to regulatory flexibility, the enhanced understanding of the manufacturing
process would allow more informed risk assessment as per ICH Q9 regarding the affects
of process changes and manufacturing deviations (excursions) on product quality.
Nice bro this content is very use ful
ReplyDeleteThanks....
Delete